Nanoscale Flow Research Division
Quantum Nanoscale Flow Systems Laboratory

ProfessorTakashi Tokumasu
It is necessary to use the method which consider the uncertainty of position of hydrogen atoms to reproduce the flow characteristics of liquid hydrogen accurately by molecular dynamics method. In this study we treat the quantum characteristics of hydrogen by using path integral centroid molecular dynamics method and analyze the mechanism by which the quantum characteristics of hydrogen affects on the macroscopic flow phenomena of liquid hydrogen.
【Novel Battery Nanoscale Flow Concurrent Laboratory】
Quantum/molecular dynamics studies of transport phenomena of substances in polymer electrolyte fuel cell
Quantum/molecular dynamics studies of transport phenomena of Li ions in all solid state battery
Construction of proton hopping model to analyze the transport phenomena of proton
In water a proton (H+) exists as an oxonium ion (H2O) by connecting with water molecule (H2O). The diffusivity of oxonium ion is 4-5 times larger than that of water molecule by the mechanism which uses a kind of chemical reaction called “proton hopping”. In this study we investigate the characteristics of “proton hopping” by various quantum calculations and make a model to treat proton hopping in the framework of classical molecular dynamics.

Reactive Force-Field Molecular Dynamics Study of deposition mechanism on the atomic layer deposition and chemical vapor deposition methods
In the semiconductor manufacturing process, there are thin film formation, etching, cleaning, etc. The atomic layer level control of film thickness error ± 0.5 Å is required on a wafer for the thin film formation process. Chemical vapor deposition (CVD) and atomic layer deposition (ALD) are widely used as thin film formation methods for realizing such a state-of-the-art demand. However, these thin film formation phenomena are a complex combination of diffusion phenomena and reaction phenomena, and it is not easy to understand in detail. Therefore, we carry out reactive force field molecular dynamics (ReaxFF MD) simulations and aim at a universal understanding of the deposition mechanism in this research.
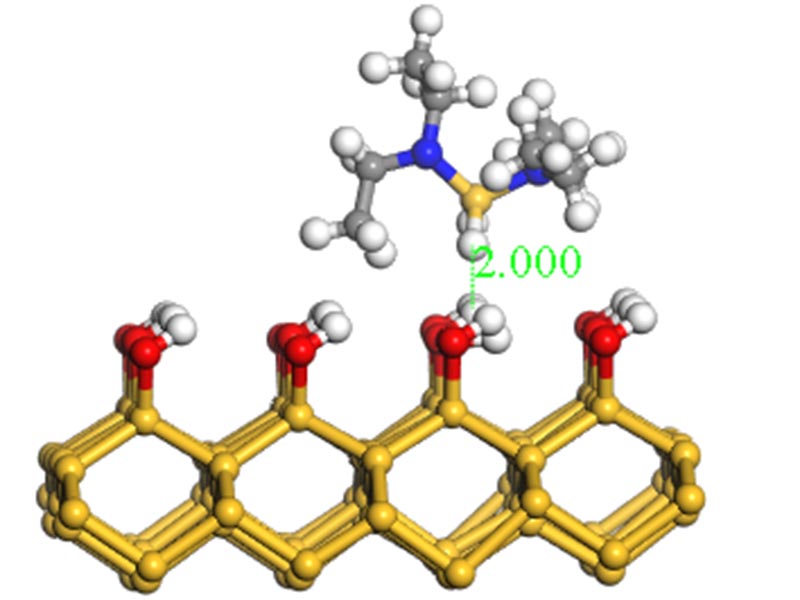 Quantum chemistry simulations for ALD processes using Bis(diethylamino)silane.
Quantum chemistry simulations for ALD processes using Bis(diethylamino)silane.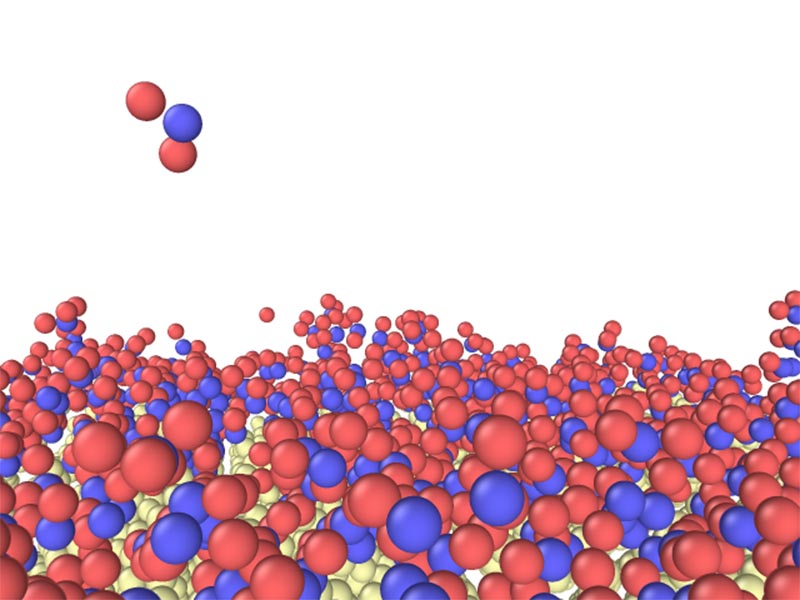 Reactive force-field molecular dynamics simulations for CVD processes using silane type precursor.
Reactive force-field molecular dynamics simulations for CVD processes using silane type precursor.
Molecular Study for Controlling Intracellular Protein Function Using Artificial Phase-Separated Structures and Selective Transmembrane Ion Channels
The research involves theoretical and computer simulation studies of biomolecular systems. Current research activities span both development of new computational methods and theoretical characterization of proton transport and protein phase behavior in biomolecular systems at multiple length scales. For example, to probe complex transport phenomena of protons, a reactive model has been developed within the simplicity of the theoretical framework of classical molecular dynamics (MD) simulations. Proton transport through complex structure such as transmembrane ion channels are one of our research interests. Protein phase behavior (i.e, aggregation, self-assembly, and liquid–liquid phase separation) in aqueous solutions are also of our research interest. Computational studies can assist in the challenge of designing the artificial ion channels. Our research is thus often carried out in close collaboration with leading experimentalists and is integrated in a feedback loop with experiments.
Click here for details >> http://mabuchigroup.fris.tohoku.ac.jp/
Click here for details >> http://mabuchigroup.fris.tohoku.ac.jp/
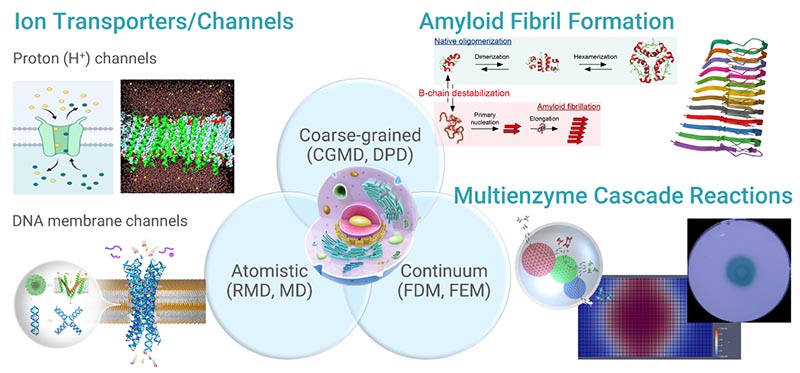 Multiscale modeling and simulation for biomolecular systems
Multiscale modeling and simulation for biomolecular systems
 【Press Release】”DIVE” into Hydrogen Storage Materials Discovery with AI Agents
【Press Release】”DIVE” into Hydrogen Storage Materials Discovery with AI Agents